Unspliced Vs Spliced Read Aligner Rna Seq
Thoughts and Theory
RNA Velocity: The Cell's Internal Compass
Finding Direction in Single-cell RNA-sequencing Data
![]()

Unmarried-jail cell RNA sequencing (scRNA-seq) has revolutionized the style we written report cell biology the by december a de, ushering in the rapidly growing field of unmarried-cell genomics. scRNA-seq allows the states to profile the transcriptome of private cells, gaining holistic insight into their function and identity. The transcriptome is the set of all messenger ribonucleic acid (mRNA) molecules of a cell. You've likely heard of mRNA frequently over this past twelvemonth with many of the COVID-19 vaccines relying on this molecule. mRNA represents the readout, or transcript, of a gene. These molecules carry data most the gene of interest that the cell's machinery uses to build a protein. In the mRNA-based COVID-nineteen vaccines, they conduct information on how to brand a benign protein tailored to the SARS-CoV2 virus, effectively teaching our torso how to fight off infection. The product of these mRNA molecules is called transcription, and these molecules are sometimes called transcripts appropriately. You'll often hear biologists refer to mRNA abundance more broadly as factor expression. You lot can remember of information technology like building IKEA furniture: the instructions are the transcribed mRNA molecule, and the corresponding protein is the furniture. Before the mRNA is translated, nevertheless, information technology has to undergo an additional step. At that place are segments of mRNA molecules that don't lawmaking for proteins called introns. These are removed prior to protein translation through a process called splicing. The remaining segments are chosen exons. This will exist important to know shortly.
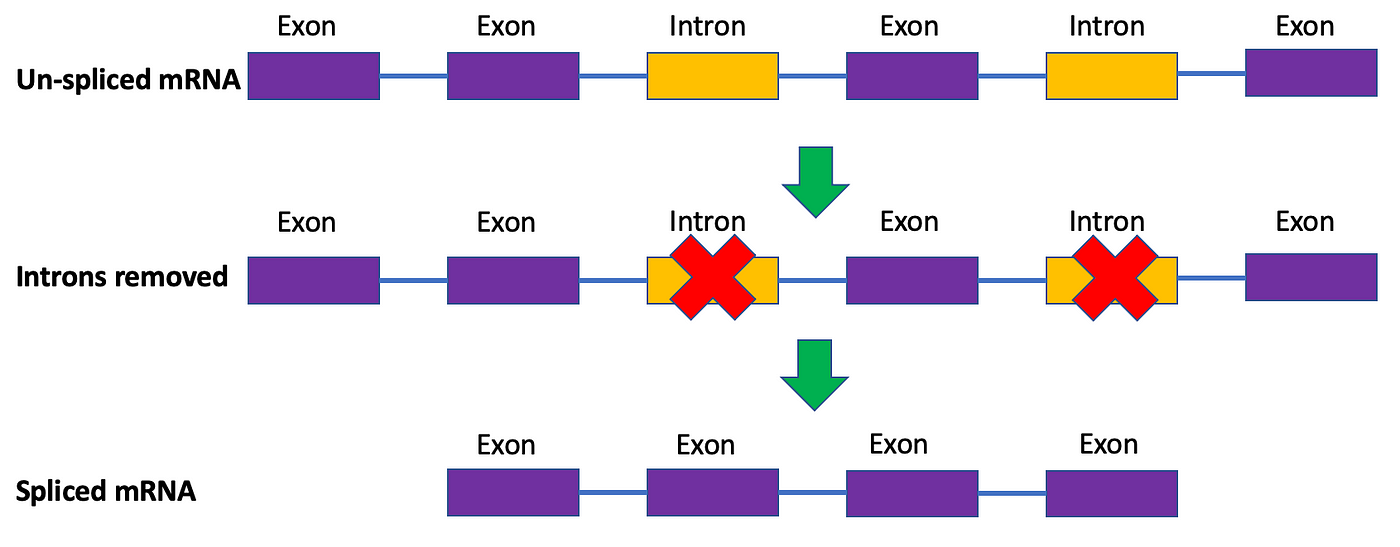
You can think of the transcriptome as the cell'south fingerprint that identifies it and distinguishes it from other cells (e.g., a red blood cell versus a neuron). By sequencing hundreds of thousands of cells for their transcriptome, we can build a holistic, loftier-dimensional map of the cellular states that stem cells traverse as they turn into more specialized cells like neurons, characterize cancer tumors, and distinguish betwixt good for you and diseased tissue from patients. These sequencing protocols consequently produce gigabytes of data to be analyzed and made sense of, which tin be complicated by having so many dimensions in our dataset, each corresponding to a gene (see my get-go article on ane mode to resolve this puzzler). Seems really cool, right? However, there is ane key drawback to this approach. These sequencing experiments kill cells in the process, preventing u.s.a. from re-sequencing these cells over again at later timepoints. Hence, we are left with a static snapshot of a jail cell's mRNA abundance, which can brand inferring its hereafter gene expression challenging. How tin can we resolve this?
This is where RNA velocity comes in. RNA velocity is a simple, all the same powerful method that allows us to predict the hereafter gene expression of a cell. Call back that I mentioned mRNA tin can be distinguished by spliced and unspliced transcripts. While spliced transcripts are the chief readout of nearly scRNA-seq protocols, unspliced expression is also measured. Using this supplemental information, we can build a simple mathematical model to predict futurity spliced expression.
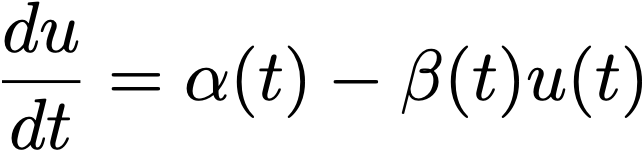
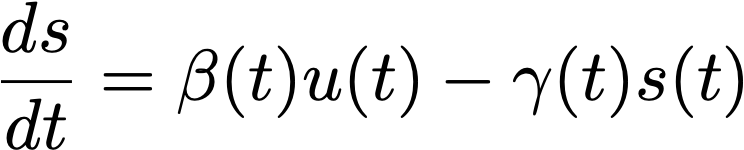
where u is the number of unspliced mRNA molecules, s is the number of spliced mRNA molecules, α is the rate of transcription, β is the rate of splicing from unspliced to spliced, and γ is the charge per unit of degradation of the spliced mRNA product.
Taking the divergence between the future mRNA expression and the current mRNA of a detail cistron for a given cell, nosotros can derive a metric of the changing gene expression. These tin can be aggregated for all genes of a given prison cell to create a vector of the cell'due south future transcriptome, representing the speed and management of said cellular modify — hence the term RNA velocity. A common way to visualize this is by superimposing a vector field onto cells in an embedding as shown below:
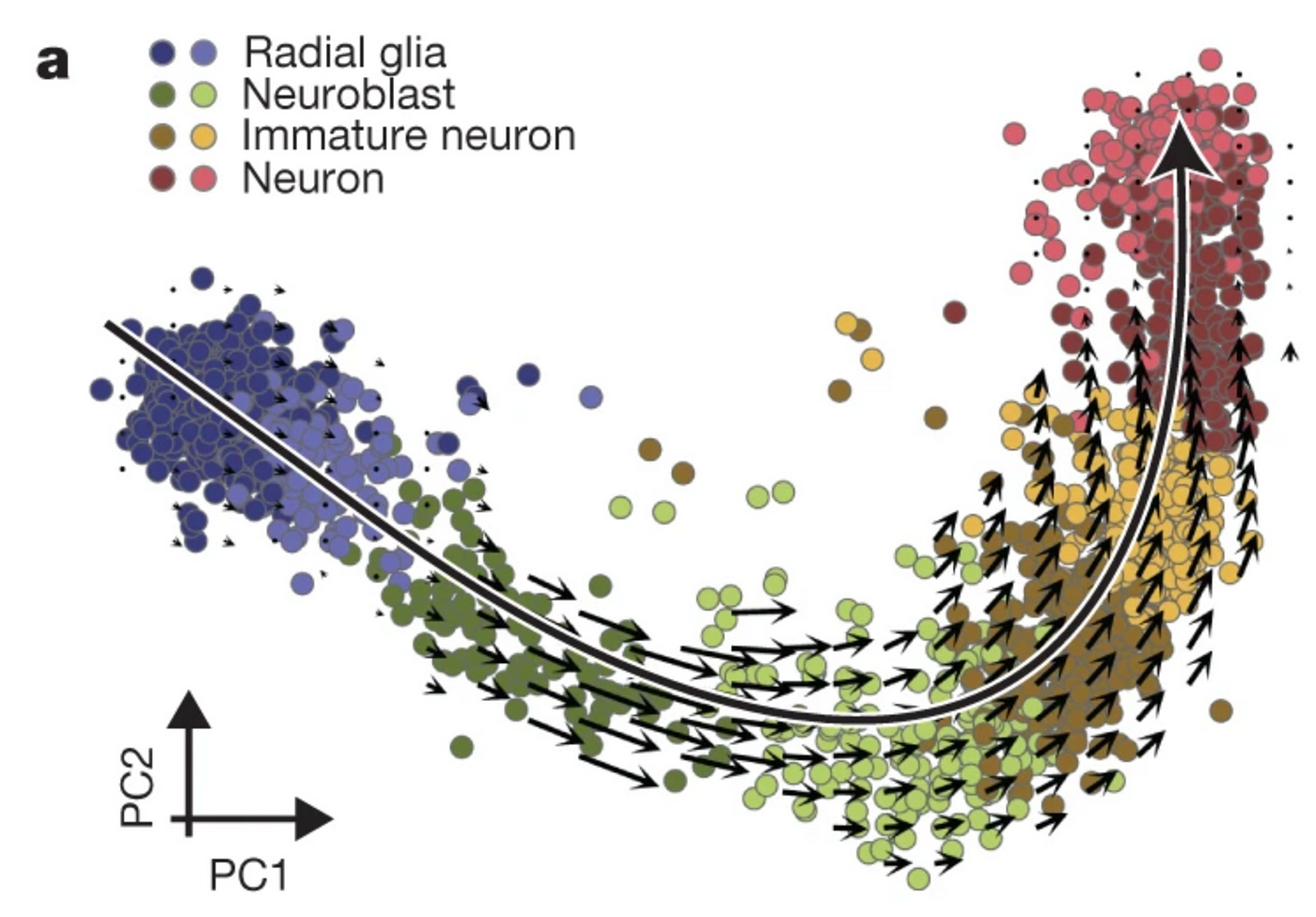
This is a PCA plot of cells undergoing neurogenesis — the production of neurons (red) in the brain from stem jail cell-like cells called radial glia (blue). Each dot represents a cell with an RNA velocity vector superimposed on it, predicting cell transitions. Notice how these arrows go longer in the intermediate stages (neuroblasts and immature neurons) — this is indicative of dynamic changes in cistron expression, as genes for neurons are turned on, while those for the radial glia cells are turned off. As these cells successfully turn into neurons, the vectors rapidly shorten, reflective of a "deceleration" of the gene expression of these cells. You tin can recall of these vectors as alike to a cell'due south compass: an internal GPS that pinpoints which direction the cell needs to get if it wants to become a neuron.
At that place are two chief models of RNA velocity: the steady-country model and the dynamical model. The steady-state model is the original model published in Nature, whose implementation is used in the to a higher place figure. This model assumes a universal, transcriptome-wide splicing rate and that factor expression follows a steady-state: i.east., velocity is estimated equally a departure from the γ-fitted ratio of spliced versus unspliced molecules when ds/dt=0 given by u = γs. I have an example of this below:
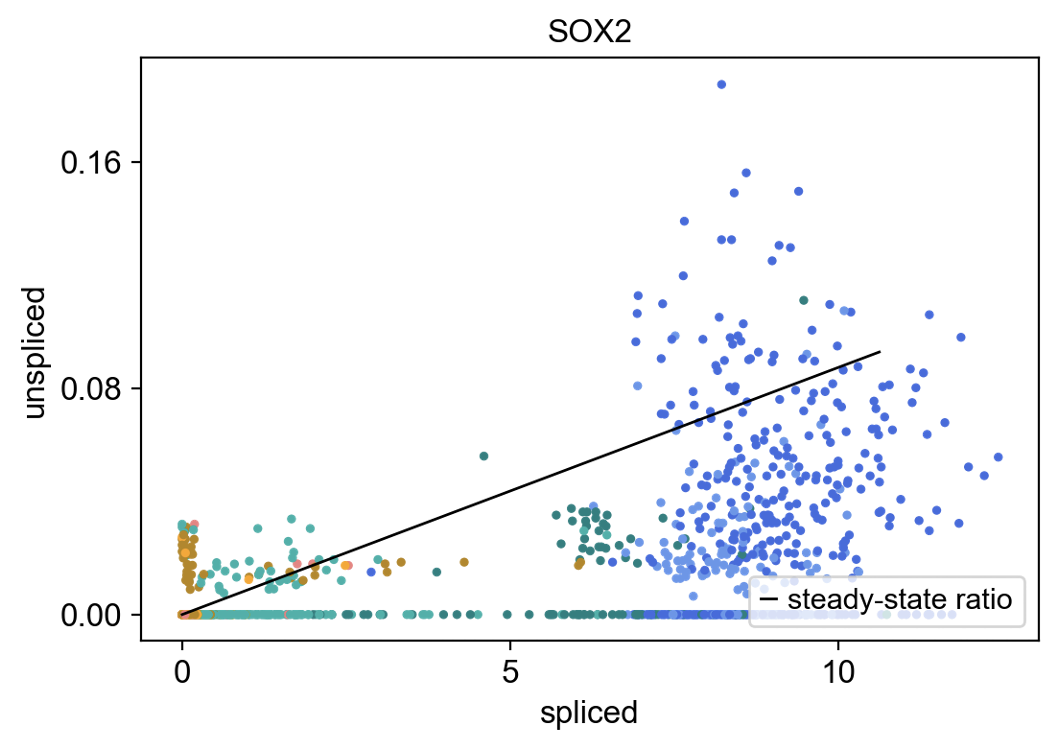
This effigy plots individual cells from the previously shown neurogenesis PCA co-ordinate to their spliced vs. unspliced molecule counts transcribed from the gene SOX2, which is a marker of radial glia cells (colored in blue). The solid line is a fit to u = γs derived from a generalized linear regression model (encounter my last article for an introduction to this statistical model). Cells in a higher place this line (u > γs)are considered to have positive velocity. This means the product of unspliced SOX2 mRNA molecules exceeds its deposition of its spliced counterpart: we have a net production of SOX2 mRNA. By contrast, cells below the line (u < γs) have negative velocity: more than SOX2 is being degraded than produced, resulting in a net loss. Cells directly on the line have a zero velocity, as they don't deviate from this γ-fitted ratio of unspliced versus spliced mRNA molecules.
The dynamical model, by contrast, direct solves for the full transcriptional kinetics for each gene, rather than making transcriptome-wide assumptions. Instead of trying to fit the data to a regression model, it estimates the parameters using the Expectation-Maximization algorithm (East-M), which uses maximum likelihood to iteratively approximate α, β, and γ, and learn the spliced/unspliced trajectory for a given gene. This assigns a likelihood as follows to each gene:
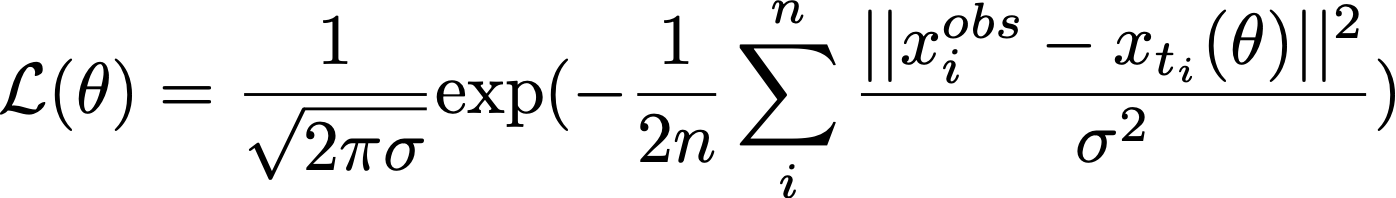
Where
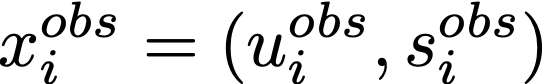
which represent the observed unspliced and spliced mRNA molecules, respectively, for a particular gene in cell i, while xₜᵢ represents the unspliced/spliced molecules at time tᵢ based on the inferred parameter prepare θ = (α, β, γ). Genes with a high likelihood are considered to be major contributors to dynamic changes in the biological phenomenon of interest — in our case, neurogenesis. Only enough math, permit'due south see this in activity!
To try out RNA velocity for yourself, install the package scvelo using the following with either pip:
pip install -U scvelo If yous prefer conda, you tin can install via the bioconda channel every bit follows:
conda install -c bioconda scvelo You volition also need scanpy, which yous can install from conda-forge. This is a popular library for single-cell assay in Python.
conda install -c conda-forge scanpy
Nosotros will use the homo neurogenesis dataset described above as an example, where I'll illustrate and compare the steady-state and dynamical models. This dataset has 1720 cells representing four main cell types in neurogenesis: radial glia, neuroblasts, immature neurons, and neurons.
First we will load the dataset from the internet:
Adjacent, let'south import our packages
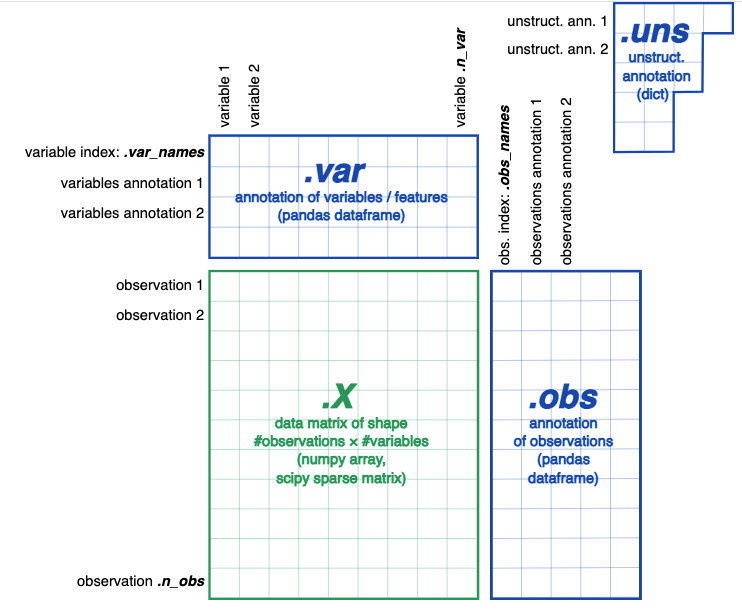
We will read our dataset into an Annotated Data (anndata) object. This a pop way to shop single-cell data for assay in Python:
Now we volition calculate the RNA velocity. scvelo uses two functions for this: scv.tl.velocity() which computes the velocities for each gene, and scv.tl.velocity_graph() which outputs a velocity graph based on cosine similarities between velocities and potential cell land transitions. In other words, it measures how well a change in factor expression of a cell matches its predicted velocity vector, which can exist used to derive transition probabilities. We'll also change the numerical cluster labels in the data to the divers cell types in the newspaper.
This gives us the following PCA plot, similar to the figure from the newspaper:
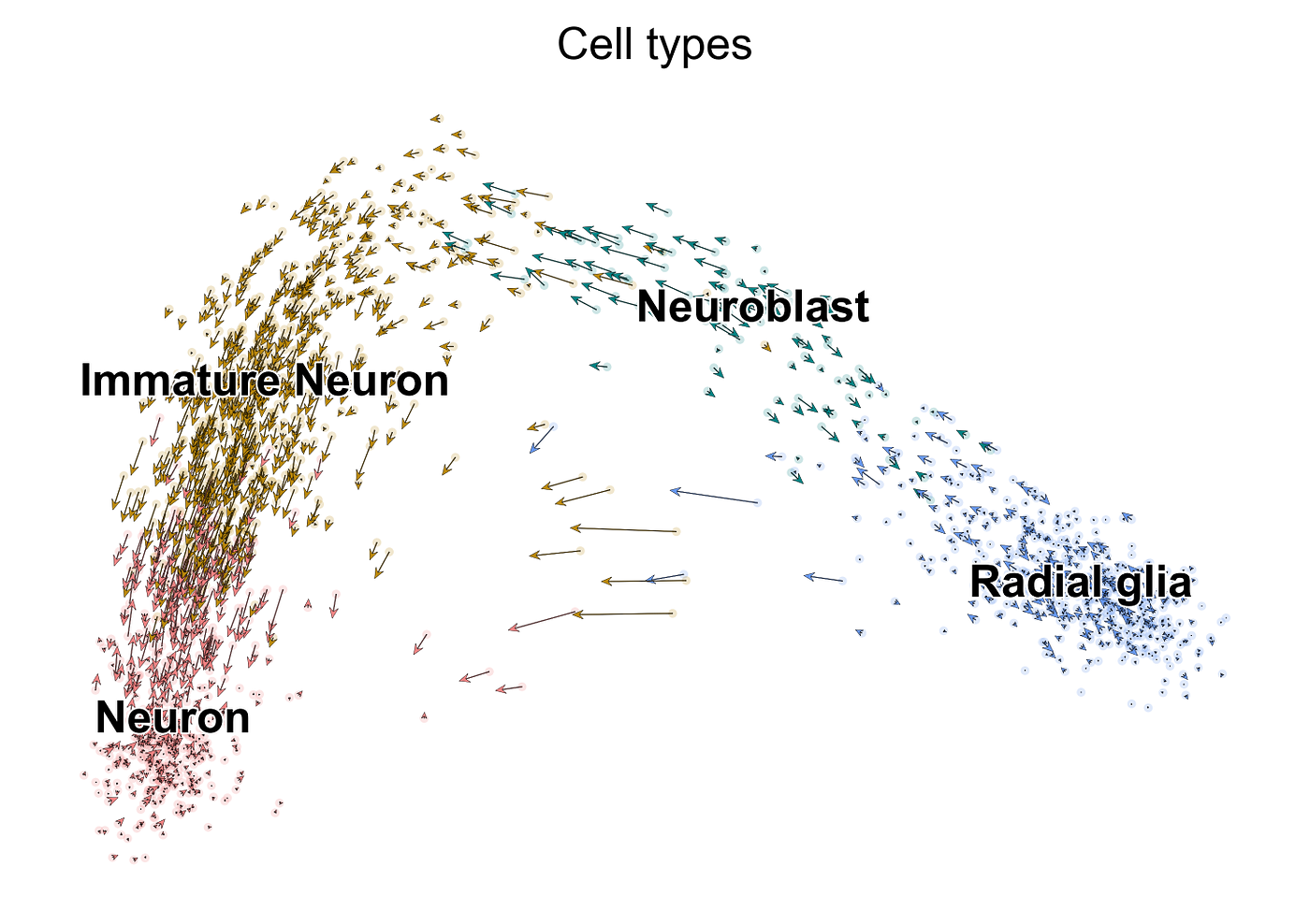
As you can run into, each cell has an arrow superimposed on it that predicts its future transcriptome. These arrows in concert give usa a sense of global directionality, showing a linear progression from radial glia cells to neurons. You may have noticed, nonetheless, that some of the neuroblast cells in green appear to be reverting dorsum to radial glia. There's similar phenomenon in some of the neurons, admitting to a lesser extent. These backflows can be attributed to two main reasons: 1) every bit mentioned previously, the steady-state assumption of the original RNA velocity model does not accurately reverberate more transient cell populations such as the neuroblasts; ii) scRNA-seq is very thin. Genes are prone to evading detection by the sequencing platform, resulting in "driblet-out", where these mRNA measurements are erroneously recorded as zeros. One mode to circumvent this is using imputation, where we computationally infer the missing values. A like approach is by data smoothing to remove the noise generated by the inherent sparsity, which the authors of the original paper did via k-nearest neighbors pooling. In that location are drawbacks to each of these methods, as well as debate as to whether they should be used at all for scRNA-seq analysis as the results tin be misleading. For the purposes of this article, I will continue present the dataset in its raw form.
We can farther explore the gene-specific velocities of cells past generating a phase portrait:
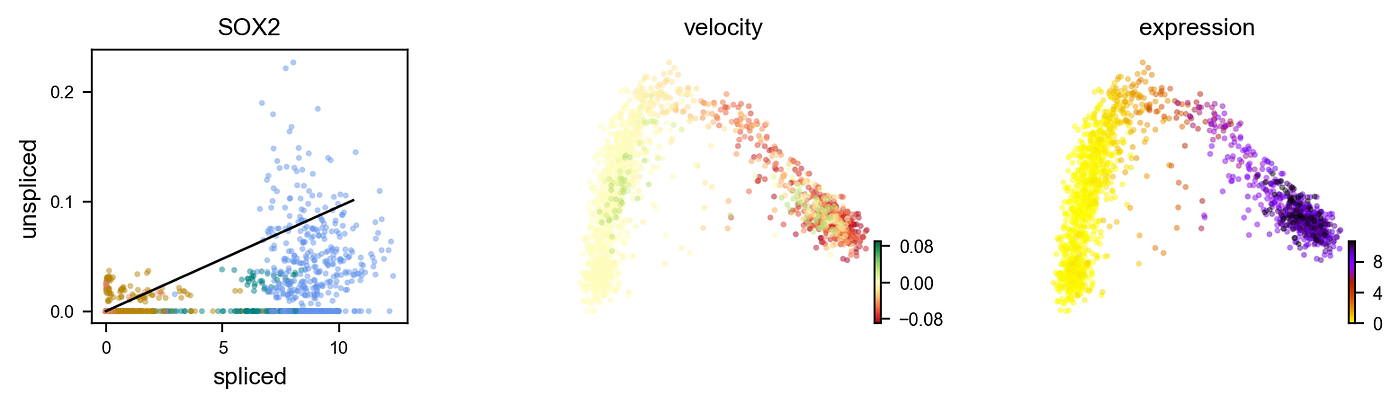
On the far left is a spliced five. unspliced plot of SOX2 expression, a marker of radial glial cells. Each point is a cell colored by its cell type (blue for radial glia, green for neuroblast, dark aureate for immature neuron, and red for neuron). Equally mentioned before, the deterministic model solves for hereafter expression by bold a steady-state model. The line represents the regression fit of the steady-land solution of ds/dt, represented by u = γs. Cells above this line are assumed to have positive velocity: SOX2 is beingness up-regulated with a net production produced in these cells more than its being degraded. We see that a few radial glia announced to display this miracle as shown by the the expression map on the correct, where cells in the above PCA plot are colored according to their levels of SOX2 expression. Nonetheless, about are shutting down SOX2 activeness (indicated by negative velocities in the centre plot) equally they begin to differentiate, as evidenced past the zero velocity of SOX2 throughout the other prison cell types: they are no longer transcribing nor degrading the mRNA molecule for it, which explains the minimal levels of expression in those cell types.
As mentioned, while powerful, the steady-state model is express in that it assumes steady-land expression levels and a constant splicing charge per unit. In more complicated systems, with multiple cease states (i.e., different cells a stem jail cell can terminally differentiate into) and heterogeneous subpopulations, this assumption can fail and yield erroneous interpretations. This is where the aforementioned dynamical model comes in. We tin can implement the dynamical model using the following code:

Here, we notice greater consensus in direction amongst neighboring cells (albeit even so a bifurcation in direction among the neuroblasts, which tin be resolved by data smoothing), with a clear dominant tendency towards neurons. Using the likelihood estimates for each factor, we tin pull out the genes that display the almost pronounced dynamic behavior:
NPAS3 0.994760
CREB5 0.896914
FOS 0.882965
SLC1A3 0.806150
SYT1 0.749349
MYT1L 0.737498
MAPT 0.687999
GLI3 0.663777
DOK5 0.656130
LINC01158 0.651856
RBFOX1 0.608533
TCF4 0.592459
FRMD4B 0.580061
HMGN3 0.577301
ZBTB20 0.575223
Name: fit_likelihood, dtype: float64 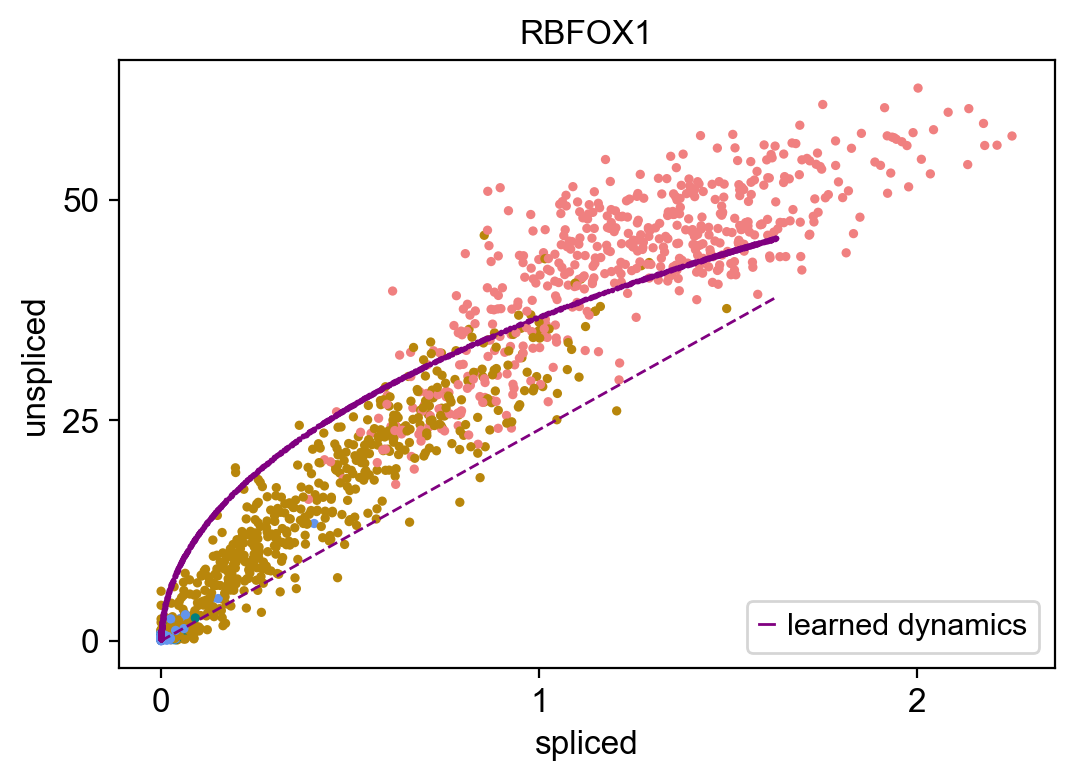
This plot shows the unspliced/spliced trajectory of the gene RBFOX1, with points colored according to cell blazon (blue for radial glia, green for neuroblast, dark gold for immature neuron, and scarlet for neuron), with a fitted bend of the learned dynamics in regal from the East-M algorithm. You lot can see how the solid curve gives a better fit than the dashed steady-state model, with a rapid increase in the transcription of unspliced mRNA relative to spliced mRNA every bit the cells get from radial glia to neurons, indicative of this gene being activated in neurons, while inactive in radial glia. We tin can observe like dynamics of several genes being up-regulated and down-regulated at dissimilar parts of neurogenesis as shown below:
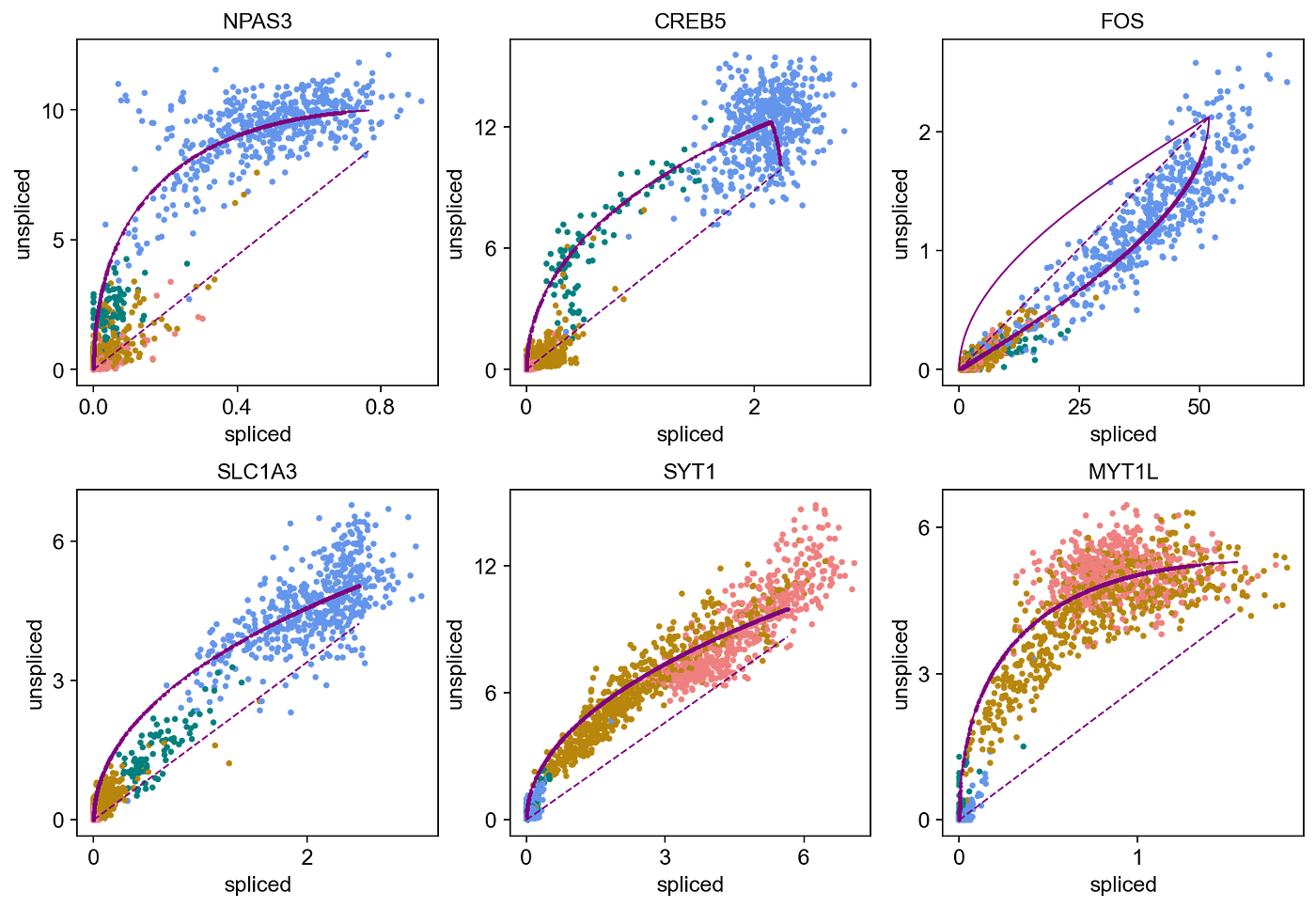
Final Thoughts
A key drawback of scRNA-seq is that it merely produces a snapshot of a cell'southward transcriptome; y'all cannot revisit your sample for successive sequencing. RNA velocity is an effective manner to circumvent this limitation by leveraging RNA biology to infer futurity gene expression, drawing out the jail cell's innate transcriptional compass and lending a sense of directionality to your data. This can be used to illustrate unlike pathways a cell may take to attain a given endpoint, how apace gene expression is irresolute, build a holistic trajectory of how stalk cells become more specialized, the list goes on. Yet, it is not without its own limitations. As mentioned before, scRNA-seq is frequently sparse and noisy. Velocities can subsequently be unintuitive or simply incorrect in the lens of basis truth for well-described biological processes. While methods like imputation and data smoothing tin can aid correct these issues, they should be used with care as they are prone to smoothing out actual biological noise that tin lend valuable insights, such as potential transitions to cells that don't be in our dataset. All that said, information technology is a powerful method that has provided a lot of clarity to making sense of high-dimensional biological information, and one that will continue to be further refined as the field of data science continues to grow. One of the goals of single-cell genomics this decade is effective integration of high-dimensional datasets measuring different properties of the cell (e.g., RNA, protein, epigenetics, etc). Methods like RNA velocity can be extended to learn the human relationship between these different modalities to make powerful inferences as to cell-to-prison cell transitions, and hence, fine-melody the cell's compass.
References:
[1] Chiliad. La Manno, R. Soldatov, A. Zeisel, Eastward. Braun, H. Hochgerner, V. Petukhov, K. Lidschreiber, M. E. Kastriti, P. Lönnerberg, A. Furlan, J. Fan, L. E. Borm, Z. Liu, D. van Bruggen, J. Guo, X. He, R. Barker, E. Sundström, Thousand. Castelo-Branco, P. Cramer, I. Adameyko, S. Linnarsson, and P. Five. Kharchenko, RNA velocity of unmarried cells, (2018), Nature
[2] Five. Bergen, K. Lange, S. Peidli, F. Alexander Wolf & F. J. Theis, Generalizing RNA velocity to transient cell states through dynamical modeling, (2020), Nature Biotechnology
[3] V. Bergen, R. Soldatov, P. V. Kharchenko, F. J. Theis, RNA velocity — current challenges and hereafter perspectives, (2021), Molecular Systems Biological science
Source: https://towardsdatascience.com/rna-velocity-the-cells-internal-compass-cf8d75bb2f89
0 Response to "Unspliced Vs Spliced Read Aligner Rna Seq"
Post a Comment